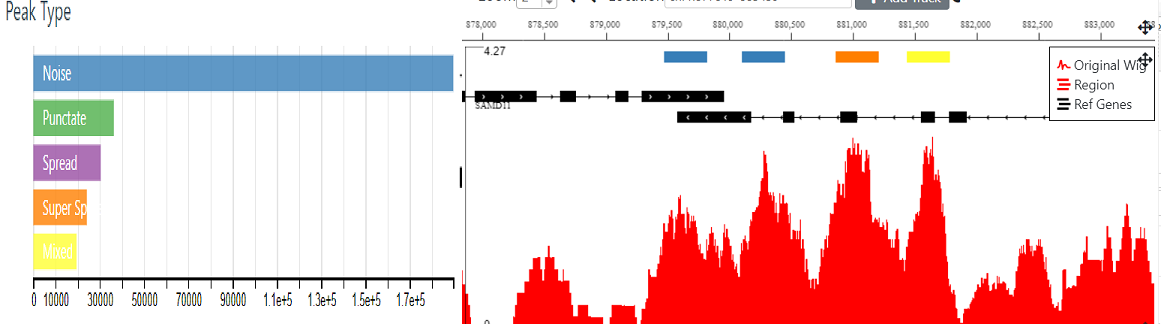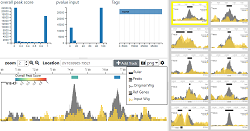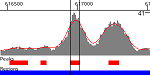
Find and Score Peaks
INPUTS:
bigWig coverage file
Find genomic locations of enriched regions using an uploaded coverage track, and score them using LanceOtron’s deep neural network